AlphaFold3が発表されたのでこれまでの技術的変遷を復習します。
初代AlphaFold (2019)
AlphaFold: Using AI for scientific discovery - Google DeepMind
インパクト: 深層学習によるゲームチェンジ
DeepMind社が2018年のタンパク質立体構造予測コンテストCASP13に提出したディープラーニングベースの構造予測モデルAlphaFoldは、未知のタンパク質構造を予測するフリーモデリングタスク(FMタスク)において2位のチームに大差をつけて1位を獲得しました。タンパク質構造予測は医薬品の開発加速に強く寄与する技術ゆえに、AlphaFoldは技術的観点だけでなくディープラーニングの産業実装という観点からも大きな関心を集めることとなりました。
手法概要
技術的な観点からは、初代AlphaFoldは①残基間距離行列の予測ステップと②二面角(ΦΨ)の最適化ステップの2ステップに分けることができます。

① 残基間距離行列の予測ステップ
このステップでは、ターゲット配列の残基数(L)×残基数(L)×特徴量数(D)の行列を入力として、220層のCNNにより残基間距離行列(正確にはCβ間距離行列)を予測します。
【入力特徴量】:
Onehot化されたアミノ酸タイプのような基本的なものからMSA由来のものまで情報がものが採用されています。
【ネットワーク構造】:
Dillated CNNが使われていること以外は特筆することがない巨大ResNetです。ちなみにL×L×D行列をそのまま入力するのはメモリがつらいので64×64×Dの行列に分割して予測したものをconcatしているようです。
【出力される距離行列】:
Cβ-Cβ間の距離行列の予測においては連続値(例: 10.3Å)を直接予測するのではなく、64binに離散化されたカテゴリを予測(例: {0: 0-1Å, 1: 1-2Å, ...., 63: 63-64Å})していることがポイントです。距離を離散化することでロス関数にクロスエントロピーを使えるため、タンパク質の大小によるスケール差の影響を受けにくい、および予測値がカテゴリ分布として得られるというメリットがあります。後者について、これにより予測分布がブロードな場合は予測の不確実性が高いというような解釈が可能になります(下図)。

② 二面角(ΦΨ)の数理最適化ステップ
タンパク質の立体構造は二面角(ΦΨ)をパラメータとして完全に表現できるので、数理最適化手法により①で予測した距離行列に合うような二面角(ΦΨ)を逆算することで実際の立体構造を作成します。
【最適化手法】:
L-BFGS法のような典型的な勾配降下法でOKとのことです。
【コスト関数V(Φ, Ψ)】:
大雑把には現在立体構造から算出される距離行列と予測距離行列の差ですが、さらにペナルティ項として立体衝突項を加えたものをコスト関数(論文中での表記はDistance potential)としています。
【初期立体構造】:
初期立体構造サンプリングのためだけにアミノ酸配列から二面角を予測するニューラルネットワークを訓練しこれを使用しています。勾配降下による最適化は初期構造に引っ張られるため、ランダムノイズを加えるなどで異なる初期構造を多数作成し、もっとも最適化結果がよかったものを最終構造として採用します。
Note: 距離行列予測は苦しい
初代AlphaFoldの主な焦点は残基間距離行列を予測することです。残基間距離行列はどのような座標の取り方をしても一意に決まるため教師ラベルとしては扱いやすいのですが、一方で矛盾のない距離行列を深層学習で生成することは容易ではありません*1。
タンパク質に限らず分子配座予測タスク一般においても2018年ごろまでは距離行列がよく使われていましたが、徐々に立体構造の直接予測にトレンドがシフトし、現在ではあまり見ることがない印象です。実際、後続のAlphaFold2では距離行列の予測は廃止され立体構造が直接予測されるようになりました。
AlphaFold2 (2021)
Highly accurate protein structure prediction with AlphaFold | Nature
過去記事:
horomary.hatenablog.com
インパクト: 驚異的な高精度と可用性
AlphaFold2は"構造生物学50年来の課題を解決"というタイトルでDeepMindブログに紹介されていますが、壮大な煽り文句に相応しいエポックメイキングな手法となっています。

まず単純に予測精度が高い(上図)。GDTスコアは90超えてれば概ね予測構造と実測構造が一致しているよねと言える指標なのですが、AlphaFold2のGDTスコア中央値は87.0であり、これは予測したらだいたい正しいという従来からは考えられないほどの驚くべき精度です。
もう一つのインパクトは可用性です。DeepMindはGoogle ColabをインターフェースとしたAlphaFold2による立体構造予測を全世界に公開したため、誰もが自身の研究にAlphaFold2を使える状態となりました。
手法概要
AlphaFold2は初代AlphaFoldから大きくアーキテクチャを変更しています。
重要な変更点として、初代では残基ペア間距離行列を出力することで間接的に立体構造を予測していたのに対しAlphaFold2では各残基の3D位置を出力することで立体構造を直接予測します。特徴抽出ネットワークについてはCNNを完全廃止し、Transformerベースのタンパク質向け特製アーキテクチャ(Evoformer)を採用したことで効果的な特徴抽出を可能としています。
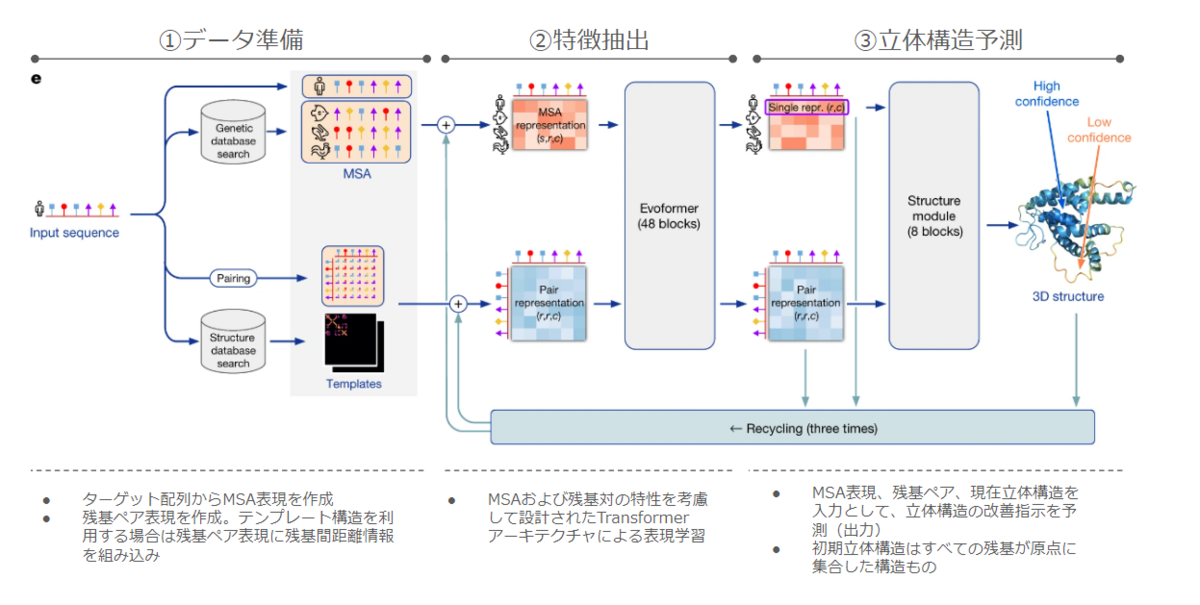
① データ準備
初代AlphafoldではMSA由来の情報は残基ペア表現行列にまとめられていましたがAlphaFold2では明示的に分離して扱うため、データ準備ステップにおいてMSA行列と残基ペア行列の2つの行列作成を行います。このステップでは各種バイオインフォツールを使用して入力データとなる行列を準備するだけでありニューラルネットは使用されないことに留意ください。

【MSA行列の作成】:
なにも特別なことはなく、バイオインフォの標準ツールであるjackhmmerやHHBlitsを使用してターゲット配列からMSA行列を作成します。
【残基ペア行列の作成】
残基iと残基j間の関係性を表現する行列を作成します。最低限の特徴量として残基i、残基jそれぞれのアミノ酸タイプが特徴量として入力されます。
オプションとしてホモロジーモデリング的な発想により、DBから検索した類似配列の立体構造の残基間距離を特徴量として使用することもできますがこれはやらなくても(ゼロ入力でも)精度が大して変わらないようです。DBから検索した立体構造を使わない場合は、すべての残基が原点に集合した構造を初期立体構造として残基間距離を計算するため、ゼロ行列が距離特徴量として入力されます*2。
② Evoformerによる特徴抽出
このステップでは、トランスフォーマーベースのアーキテクチャであるEvoFormerを用いてMSA表現および残基ペア表現の特徴抽出(表現学習)を行います。Evoformerは構造生物学のドメイン知識を取り入れた設計になっていることが興味深いポイントです。

【MSA行列の表現学習】
MSA表現の特徴抽出ネットワークではaxial self-attention(軸方向アテンション)を用いて配列方向(横)とアライメント方向(縦)の両方向から情報を読み取り、MSA表現に追記するというような操作を行います。
MSAを縦横に見るというプロセスは人間がMSAを読むときと同様であると解釈できます。例えば、横(配列方向)に見ることで、「このあたりの配列はαヘリックスっぽいな」という情報をMSAに追記し、横(アライメント方向)に見ることで、「変異がほぼないので構造的に重要な部位に違いない」というような情報をMSAに追記することを繰り返しているようなイメージです。
【残基ペア行列の表現学習】
初代Alphafoldでは残基ペア行列からの特徴抽出(表現学習)はCNNベースのResNetで行っていましたが、AlphaFold2では完全にトランスフォーマーに置き換えられています。なお、残基ペア行列の表現学習ネットワークはタンパク質の空間的制約を考慮し、残基i-j間の特徴更新では残基i, jにかかわるすべての残基ペア特徴(i-k, j-k)が入力されるネットワーク構造を採用しています。直感的には、このような設計にすることで残基i-残基jの距離情報の更新においてほかの残基との位置関係情報が考慮されやすくなるのだろうと解釈できます。
【MSA表現と残基ペア表現との情報交換】
EvoformerにおいてMSA表現は定期的(1blockごとに1回)に残基ペア表現に情報提供を行うような構造になっています。直感的には、例えばMSAから「この部分の配列はほぼβシートだろう」という情報を獲得できた場合、これを残基ペア表現に反映することで「βシートなら残基間距離はこんな感じになるよね」という情報を追加していると解釈することができます。
③ 残基ベース立体構造予測
このステップではMSA表現、残基ペア表現、現在の立体構造を入力として、より改善されたタンパク質立体構造を予測します。立体構造予測は”主鎖レベルの位置決め”と”側鎖レベルでの位置決め”の2段階で行われます。

【主鎖レベルの位置決め】
IPA module (Invariant Point Attention) では、MSA表現、残基ペア表現、現在立体構造を入力として、より改善された立体構造とするための各残基への相対的な並進・回転指示を出力します。各残基への相対的な並進・回転指示とは具体的には例えば、残基番号1のリシンは右に3歩移動(並進)して向きを上方向に30°変えて(回転)、残基番号2のアラニンは...、というような指示をイメージしてください。このような相対移動予測による立体構造改善を何度も繰り返すことで最終立体構造を取得します。
このようにAlphafold2では初期立体構造(原点にすべての残基が集合した構造)からワンショットで最終構造を予測するのではなく、予測と改善を繰り返して最終構造を取得するような仕組みになっています。これはタンパク質立体構造予測という困難な大問題をより簡単な部分問題に分割することで解きやすくしていると解釈できます。直感的には、一発書きでお絵描きするよりラフ→線入れ→塗りとステップを分けて絵を描いたほうが容易になるイメージ。
【側鎖レベルでの位置決め】
残基粒度での位置決めに引き続き、側鎖のねじれ角を予測することで側鎖構造レベルでの位置決めを行います。
Note: SE(3) 同変な構造予測モデル
タンパク質に限らず3D空間におけるグラフや点群の生成問題においては、特別なネットワークを使用しない限りは入力点群の座標の取り方によって出力構造予測も変化してしまうことが問題になります。入力構造の向きを変えたら(Pymolで分子をグルグル回すイメージ)出力値も変わってしまうというのは不安定で好ましくないですし、そもそも学習効率が悪いです。
そこで構造予測を担うIPAモジュールでは、入力構造については座標の取り方に影響を受けない残基間距離を特徴量とすることで、出力(各残基への相対的な移動指示)も入力構造の並進・回転の影響を受けないような設計となっています。*3
AlphaFold3 (2024)
インパクト: 折り畳みから分子間相互作用へ
AlphaFold3ではタンパク質と生体分子(低分子化合物/修飾残基/DNA/RNA/タンパク質)との複合体構造の予測に焦点を当てることで実用性を大きく高めています。 これはたとえば医薬品開発においてはタンパク質立体構造そのものよりかは、受容体と低分子リガンド、タンパク質と核酸、抗体と抗原などのような分子間相互作用の解明こそが重要であるためです。

AlphaFold2までの立体構造予測アーキテクチャはあくまでアミノ酸残基が主役であり、低分子化合物やDNA/RNAとの複合体構造を予測することが仕組み上不可能でした *4。このため、AlphaFold3では立体構造予測のアーキテクチャをAlaphfFold2から大きく変更し、アミノ酸残基粒度でなく全原子粒度で立体構造予測を行うことで全原子粒度での構造予測=分子間相互作用を含めた構造予測を実現しています。

とくに印象的なのがPoseBusterドッキングベンチマーク結果を示したExtended Data fig.4(上図)。結合ポケット位置の指定なしでも70%強の正解率、結合ポケット周辺の残基を指定した場合の正解率は80%オーバーでこれは代表的な古典ドッキングツールGoldよりも良好な結果であり、AlphaFold3は低分子ドッキングツールとしても最先端の精度を発揮するという結果を示しています。
①全原子拡散モデルによる分子構造生成
近年の深層学習による分子立体構造生成は画像生成分野で急速に発展した拡散モデル(Diffusion model)を利用した手法が主流となっていることから、AlphaFold3では拡散モデルベースの立体構造予測アーキテクチャが採用されました。
拡散モデルの有用な特性の一つは高精度な条件付け生成が可能であることです。たとえばStable Diffusionのような画像生成サービスでは、ユーザーの入力テキストで拡散モデルによる生成を条件づけることで任意の画像生成を実現します。AlphaFold3も同様にタンパク質配列および周囲の生体分子情報で拡散モデルによる生成を条件づけることによって任意の分子立体構造生成を実現します。
ゆえにAlphafold3とは長大な条件付けネットワークを備えた全原子拡散生成モデルであると表現できます。

全原子拡散生成モデルなのでシンプルに原子座標だけを予測します、すなわちガウスノイズの乗った原子数分のxyz座標を入力としノイズの低減された原子数分のxyz座標を出力します。結合情報は不要です、なぜなら拡散モデルが予測した原子間座標から原子間距離を計算すれば単結合なのか、二重三重結合なのか、それとも結合していないのかを判定できるからです。つい忘れがちですが分子はボール&スティックではないのです。
ちなみに、従来の全原子拡散モデルによる分子生成研究(EDMとか GeoDiffとか )においてはいかに立体構造生成モデルにSE(3)同変性を導入するかが主要な論点であり、AlphaFold2においても立体構造予測がSE(3)同変になるように工夫を凝らしていたのですが、
最近のいくつかの研究と同様に、分子のグローバルな回転と移動に関する不変性や同変性はアーキテクチャでは必要ないことがわかったため、機械学習アーキテクチャを簡素化するためにそれらを省略しました。(機械翻訳)
とばっさり切り捨てられているのが驚き。
分子生成におけるSE(3)同変性については過去記事を参照:
horomary.hatenablog.com
② 長大な条件付けネットワーク
Alphafold3とは長大な条件付けネットワークを備えた全原子拡散生成モデルであると前述したとおり、Alphafold3では必須入力としてタンパク質配列を、任意入力として核酸配列、SMILES形式で表現された低分子リガンド、金属イオンなどを長大な条件付けネットワークに入力することで、拡散モデルへの条件付けベクトルを作成します。

条件付けネットワークにおいて、InputEmbedder部についてはアミノ酸以外(DNA/RNAや低分子リガンド)にも効率よく対応するためにLLMのようなトークン化機構が備えられています。すなわち、タンパク質、DNAやRNAのように構成要素が決まっている分子については残基単位でトークン化し、そうでない低分子リガンドや残基修飾、金属イオンについては原子単位でトークン化することで計算量を減らしつつ汎用性を確保します。それ以外はAlphafold2のEvoformerを全体的に単純化したものという印象。
③ 相互作用を重視した蒸留データセット
RNAやDNAとの相互作用を含むタンパク質結晶構造は少なく、PDBデータセットから単純に学習するだけではAlphaFold3の焦点である分子間相互作用を学習することが困難であるため、AlphaFold2、AlphaFold2-Multimer、AlphaFold3による予測構造を教師データに含めるとすることでデータの少なさを補います。なお、このような学習済みモデルによる予測データを教師データに含めて新たなモデルを学習する手法を蒸留(distillation)と言います。

興味深いのはdisorder領域(無秩序領域)を含む構造データセットをAlphaFold2予測により作成している点です。どうやらAlphaFold3の拡散モデルベース予測では本来かっちりとした構造をとらない無秩序領域であっても何かしらのモチーフを取りたがる傾向があるようで、この弱点をAlphaFold2による蒸留データセットで補完しています。
無秩序領域は分布としてはノイズに近いだろうために、デノイジングフィルターたる拡散モデルが何かしらのモチーフ取りたがらせるのはまあそりゃそうだろうなといったところ。
AlphaFold4(いつかな?)
注: 以下は単なる個人の想像であることに注意してください。24年6月現在AlphaFold4についての公式発表はありません。
次は構造分布の予測?
AlphaFold1,2では高精度なタンパク質立体構造予測を実現し、AlphaFold3では高精度なタンパク質-生体分子複合体構造、すなわち分子間相互作用の予測を実現しました。残された大きな課題はタンパク質立体構造分布の予測であるように思います。
タンパク質構造予測モデルの主な制限は、通常、PDB に見られるような静的構造を予測し、溶液中の生体分子システムの動的動作を予測しないことです。この制限は AF3 でも変わりません。AF3 では、拡散ヘッドまたはネットワーク全体の複数のランダム シードでは、ソリューション アンサンブルの近似値は生成されません。(AF3論文より機械翻訳)
拡散モデル(というかDenosing Score Matching)はボルツマン分布を仮定した生成モデルのため(詳しくは 拡散モデル データ生成技術の数理を読もう)、シードを変えて繰り返し生成したら溶液中の構造分布を再現するのでは?と期待してしまいます。しかし、論文内でも言及されているようにAlphaFold3はあくまでPDBの静的構造を予測するように訓練されており立体構造分布を再現することはありません。
たとえば、E3 ユビキチンリガーゼは、アポ状態では自然に開いた立体構造をとり、リガンドに結合したときに閉じた状態でのみ観察されますが、AF3 はホロシステムとアポシステムの両方で閉じた状態のみを予測します(AF3論文より機械翻訳)
もしタンパク質立体構造分布が予測できるようになると結合サイトのダイナミクスに合わせた薬剤設計が可能になるなど多くの嬉しさがあります。
さらに、タンパク質-生体分子複合体の立体構造分布を予測することができるならばそれは結合自由エネルギーを予測できることと同義であるので創薬において非常に大きな意味を持ちます。AlphaFold3ではタンパク質と生体分子がどのように複合体形成するかまでしか推定することができませんが*5、複合体立体構造分布が予測できるとその複合体の結合強度がどの程度かまで推定することができます。
拡散モデルによる構造分布予測
DeepLearningで大規模分子の構造分布を予測するなんて数年前には考えられませんでしたが、拡散モデルによってすでに現実になりつつあります。一例として Distributional GraphormerというMicrosoft Researchの研究を紹介します。

この論文では拡散モデルを含む深層学習によって分子システムの平衡分布を予測する方法を提案しています。具体的にはMDシミュレーションで得られた構造分布を教師データとして拡散モデルをトレーニングすることで、アデニル酸キナーゼや合金の表面触媒系など複数のタスクにおいて構造ランドスケープを生成的に再現することに成功しています。これは創薬だけでなく素材化学においてもインパクトの大きい結果です。
もし、このような拡散モデルによる構造分布予測手法をAlphaFoldに組み込むことができればAF3論文内で言及されていたlimitationも解決することができ、ますます実用性が高まります。
たとえば、E3 ユビキチンリガーゼは、アポ状態では自然に開いた立体構造をとり、リガンドに結合したときに閉じた状態でのみ観察されますが、AF3 はホロシステムとアポシステムの両方で閉じた状態のみを予測します(機械翻訳)
統計力学と相性の良い拡散モデルを導入したこともあり、このような構造分布の再現というのはAlphaFoldの発展の方向としてありうる可能性ではないでしょうか。(知らんけど!)
参考書籍
現代化学 2024年7月号 AlphaFold3,その正体を探る
*1:距離行列のつらさは https://arxiv.org/abs/2105.03902 が詳しい
*2:実際は初代AlphaFoldと同様に連続値は離散化されるのでゼロ行列ではないがわかりやすさのため
*3:より正確にはIPAモジュールによるsingle representationの更新はSE(3)不変操作だが、各残基への相対的な移動指示はSE(3)同変な操作
*4:タンパク複合体の予測も意図されたものではなく、某先生がハッキング的な使い方を発見したことによる。ちなみにこの先生のブログには以前に分子動力学やってたとき大変お世話になった思い出があり感慨深い。
*5:もちろんタンパク質-低分子リガンドくらいなら既存のドッキングスコア関数である程度は結合強度を推定することができる