注意:
- Alphafold2の手法解説です。使い方の説明ではありません
- 構造生物学ドメインにはある程度の説明をつけます
- アーキテクチャ設計の意図については個人の考察であり、正しさに何ら保証がありません
- AttentionとTransformerそのものについての説明は行いません
- AlphaFold2とは
- タンパク質折り畳み問題について
- AlphaFold2の概観
- 0. データ準備
- 1. Embeddingモジュール
- 2. Evoformerモジュール
- 3. 構造モジュール (Structure module)
- ロス関数
- 「予測の信頼性」の予測:pLDDTスコア
- 更なる精度向上のためのトリック
- まとめ
- 補足情報
AlphaFold2論文など:
Highly accurate protein structure prediction with AlphaFold | Nature
Supplementary information(アルゴリズム詳細説明)
AlphaFold: a solution to a 50-year-old grand challenge in biology | DeepMind
AlphaFold2とは
タンパク質は生命に不可欠であり、実質的にすべての機能をサポートしています。タンパク質はアミノ酸の鎖で構成された大きな複雑な分子であり、タンパク質が何をするかはその独特の3D構造に大きく依存します。タンパク質がどのような形に折りたたまれるのかを解明することは「タンパク質の折り畳み問題」として知られており、過去50年間生物学の大きな課題となっていました。このたび、AIシステムAlphaFoldの最新バージョンは、隔年で開催されるタンパク質構造精密予測コンテスト(CASP)の主催者によってこの壮大な課題の解決策として認められました。この画期的な進歩は、AIが科学的発見に与える影響と、私たちの世界を説明し形作る最も基本的な分野のいくつかで進歩を劇的に加速する可能性を示しています。(DeepMind Blog より)
DeepMind社のタンパク質の立体構造予測アルゴリズムAlphaFoldの最新版である「AlphaFold2」がCASP14(タンパク質立体構造予測コンペ)にてエポックメイキングな結果を残しました。とくに構造未知タンパク質の立体構造*1を予測するという非常に難しいタスクでも、GDT(ざっくり正しい位置を予測できている原子の割合)が87.0という驚くべき精度となっています。

今後AlphaFold2は構造生物学的研究の必須ツールの一つとなっていくことが予想されますが、内部の仕組みがよくわからないツールを使うのはいくら精度が高いことが実証されていても心地悪いものです。そこで、本稿ではこの「AlphaFold2」のタンパク質立体構造予測の仕組みを論文に基づいて解説していきます。
タンパク質折り畳み問題について
まずは前提知識としてタンパク質折り畳み問題の概略を説明します。※生物系の方はスキップ推奨
タンパク質はバイオ・ナノマシン
タンパク質とはアミノ酸が鎖状に連結した高分子化合物でありすべての生命に不可欠な物質です。タンパク質は筋肉を動かすモーターとして働いたり、赤血球として全身に酸素を運んだり、あるいは緑色に光ることさえできます。生命活動に必要なほとんどすべての機能発現がタンパク質によって担われているのです。

・タンパク質は20種類のアミノ酸で構成される鎖
多様な機能を発現するタンパク質ですが、驚くべきことにすべてのタンパク質はたった20種類*2のアミノ酸が鎖状に連結することによって構築されています。
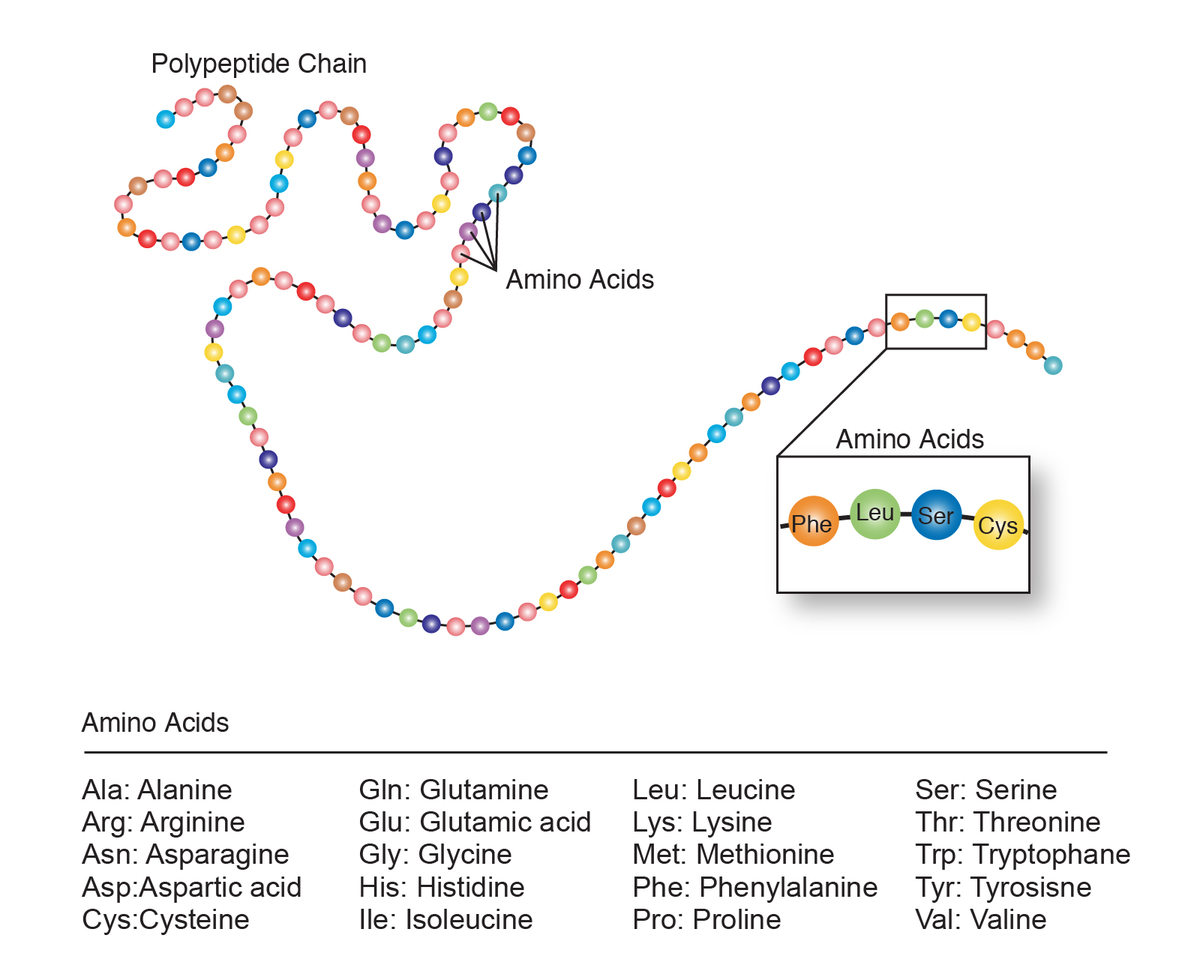
20種類のアミノ酸のうちでも、アスパラギン酸(Aspartic acid) やトリプトファン (Tryptophan)などは栄養ドリンクやサプリに表記されているのを目にしたことがあるのではないでしょうか。他にはグルタミン酸(Glutamic acid)などはうまみ調味料の主成分としてお馴染みですね。
補足資料として下図ではタンパク質を原子レベルで描画した場合の構造とタンパク質構造に関する用語をまとめました。とくに、Cα(α炭素)の位置とねじれ角(torsion angle)Φ/Ψ/ω は知らないとAlphaFold2論文を読むのが困難なので覚えておきましょう。

・タンパク質は自発的に立体構造を形成する
ここまででタンパク質はアミノ酸できた鎖(ペプチド)であることを説明しましたが、このアミノ酸の鎖は適温の水*3に入れることで自発的に*4折りたたまれて立体構造を形成します。 異なるアミノ酸配列では異なる構造に折りたたまれるためにタンパク質は多様な立体構造と多様な生物学的な機能を持つことができるのです。
補足情報:細胞内でのリアルな折り畳みについての解説
田口 英樹 「タンパク質フォールディングの「理想」と「現実」:凝集形成とシャペロンの役割」日本生化学会
タンパク質折り畳みの駆動力となっているのはタンパク質内部の分子間相互作用や水との相互作用などの物理化学的な力であるので、ごく小さいサイズのタンパク質であれば分子シミュレーション(分子動力学シミュレーション, Molecular Dynamics) によって折り畳み過程をシミュレートすることもできます。が、しかし分子シミュレーションで立体構造予測をするのは計算コストが重すぎるので現時点ではあまり実用的ではありません。
タンパク質立体構造の重要性
タンパク質の立体構造がわかることには様々な嬉しさがあるのですが、もっとも身近なのは創薬への応用です。たとえばインフルエンザ治療薬として有名なリレンザやタミフルは、ウイルスが増殖するために重要なインフルエンザノイラミニダーゼというタンパクの働きを阻害するような低分子化合物をタンパク質構造に基づいて設計することによって見出された薬です。

・ タンパク質立体構造解析の困難
問題はタンパク質の立体構造を実験的に解明するためには多大な労力が必要であることです。
もっともポピュラーなタンパク質立体構造の解析手法はX線結晶構造解析法です。この手法ではその名の通りタンパク質の結晶をX線で解析するのですが、そもそもタンパク質結晶の作製難易度が極めて高いという難点を抱えています。結晶成長に重力が悪さするから宇宙で結晶つくろうぜという実験が実際に行われている、という事実からも苦労が推し量れます。
他には NMR(核磁気共鳴法)という分析機器を使ったり、最近ではCryo-電子顕微鏡でタンパク質を直接見るというような方法も行われています。
データ駆動の立体構造予測
過去50年間にわたり構造生物学者たちは大変な労力をかけて実験的なタンパク質立体構造解析を行ってきました。このような先人の血と汗とPEGの結晶として2021年現在では18万件以上のタンパク質の立体構造がPDB(Protein Data Bank) に登録されています。
これほどのデータが蓄積されているならば教師あり学習の機運が高まるのは自然な流れです。
タンパク質の立体構造は
・ 似ているアミノ酸配列であれば同じような立体構造となる
・ 同じアミノ酸配列で同じ温度の水中なら同じ立体構造となる (と概ね見なせる)
という教師あり学習に適した特性を持っているために、データ駆動での立体構造予測の試みが古くから行われていました。
AlphaFold2ではこのような伝統的なタンパク質構造予測アプローチと深層学習をうまく融合させたことにより、圧倒的な性能を実現しました。特筆すべきはAlphaFold2で使われている深層学習のテクニックは最先端のものですが、設計コンセプト自体は伝統的なバイオインフォマティクスの発想(ドメイン知識)に基づいたものであり、所謂「ディープでポン」の対極にあるアプローチであるという事実です。
つまりはAlphaFold2は、深層学習にドメイン知識をどうやって注入すればよいのか?という視点で見ると大変示唆深く面白い手法と感じます。
AlphaFold2の概観
前提の説明が長くなりましたがここからが本題です。
4つのモジュール

AlphaFold2は4つのモジュール*5によって構成されています。
0. データ準備モジュール:
立体構造予測を行いたいアミノ酸配列(input sequence)をクエリとし、バイオインフォマティクスのツールを用いて
・ DBからのMSA(Multiple sequence alignment, 後述)作製
・ DBからのテンプレート立体構造(鋳型構造, 後述)検索
を行います。ただしテンプレート立体構造の使用は任意であり無くても構いません。
1. Embeddingモジュール:
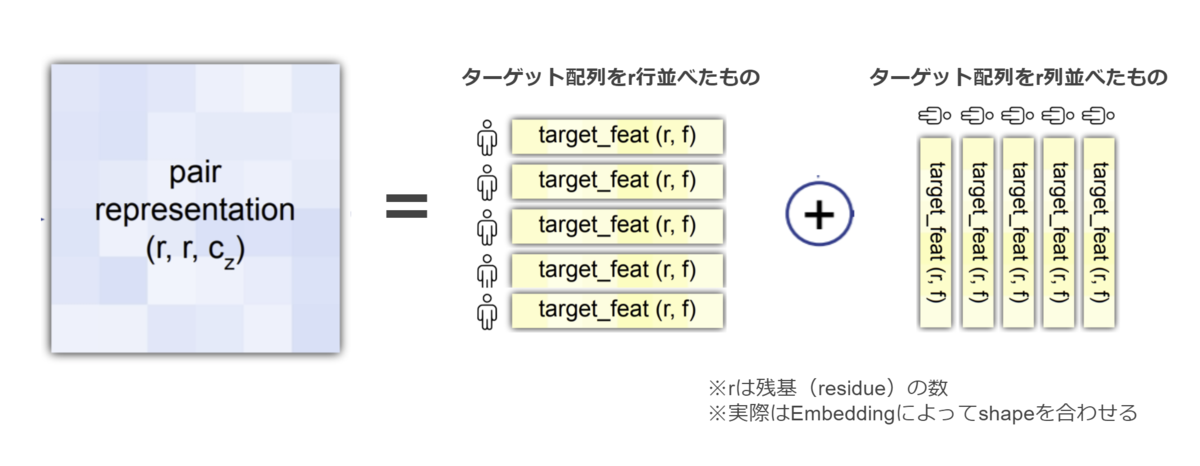
・生MSAにターゲット配列情報を紐づけた MSA representation の作成
・残基間の相対的な位置関係を記録する Pair representation の作成
・スパースな入力値に対してEmbedding(活性化なし全結合層)を行いdenseなベクトルに変換

2. Evoformerモジュール:
Evoformer=Evolution(分子進化)のためのTransformer
・MSAからの特徴抽出
・Pair representationからの特徴抽出
・MSAとPair representationでの情報交換
やりたいことはTransformerのEncoder部とほぼ同じですが、MSAの特性や空間グラフ(タンパク質)の物理的な制約を意識してちょっと変わったattentionの設計(axial attention, triangular attention)が行われています。

3. 構造モジュール:
・IPA (Invariant point attention)モジュールによるMSA表現, 残基ペア表現そして現在の立体構造の統合
・各残基への相対的な移動指示(=残基数分の(3,3)の回転行列と(x, y, z)の並進ベクトル)および側鎖のねじれ角(χ1-4)を予測
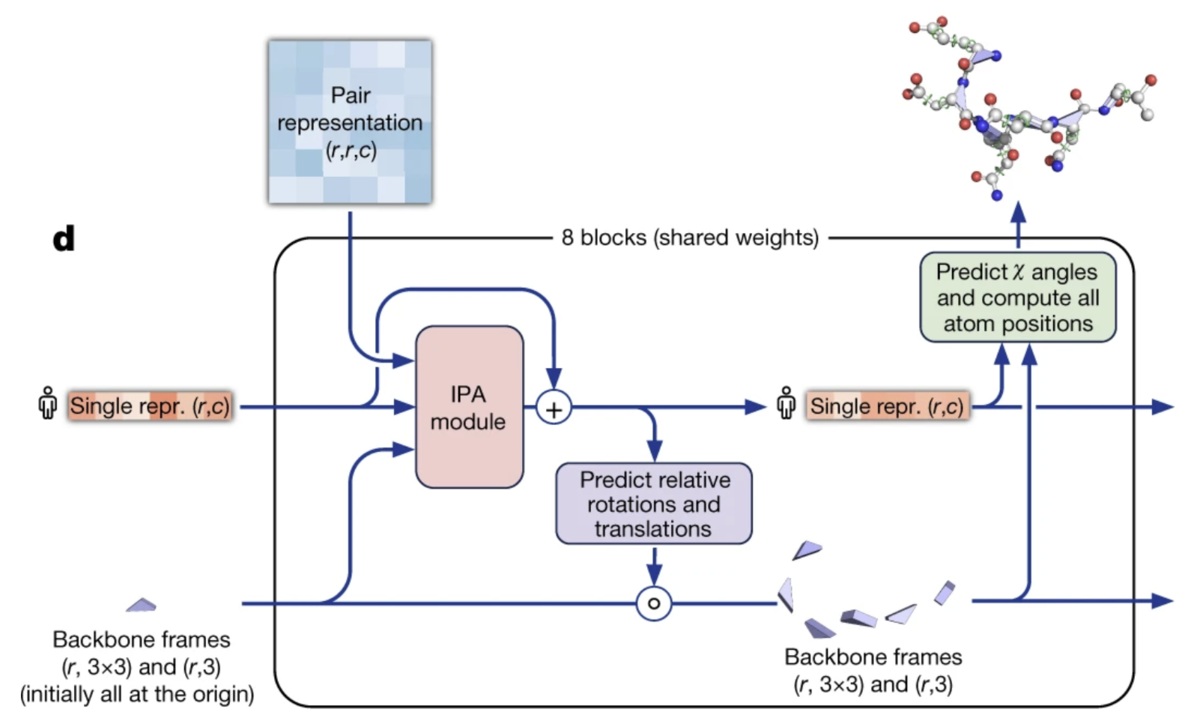
AF2のやってることをざっくり理解する
AlphaFold2とはタンパク質立体構造を入力としてよりrefineされたタンパク質立体構造を出力するネットワークであると理解できます。
AF2の処理を極めて単純化すると以下のようになります。
① すべての残基が原点に集合している構造で主鎖立体構造を初期化(ブラックホール初期化)
② 現在の主鎖立体構造とMSA表現およびペア表現を構造モジュールに入力し、各残基への相対的な移動指示を出力
③ 現在の主鎖立体構造に②の出力を適用し、主鎖立体構造を更新する
④ 2-3を一定回数繰り返すことで最終的な立体構造を得る

(実際には側鎖のねじれ角も予測しますが)おおまかな理解としてはAlphaFold2は各残基ごとへの相対的な移動指示(回転と並進)の出力を繰り返すことによってより良い立体構造を模索しているイメージとなります。ここで、相対的な移動指示というのは「残基番号127のグルタミン酸は今の位置から10歩くらい右に動いて」というようなニュアンスです。
ツールとしてのAlphaFold2ユーザーの視点では目的配列を入力すると一発で精確な立体構造が出力されるように見えますが、実際には分子力学シミュレーションによる構造最適化と同じように相対的な立体構造改善のiterationを重ねることで最終的な立体構造を得ています。構造モジュール(Structure module)はこのような立体構造改善iterationを実行する役割を担っています。
また、AlphaFold2が立体構造改善の重要な手掛かりとしているのがMSA(Multiple sequence alignment)です。後述しますがこれはMSAにはアミノ酸配列-タンパク質立体構造相関情報が豊富に含まれているためです。EvoformerモジュールはMSAからの情報抽出を担っています。
0. データ準備
伝統的なバイオインフォマティクス手法による入力データ準備を行うモジュール*6です。立体構造を予測したいタンパク質のアミノ酸配列 (input sequence)をクエリとしたDB検索によってMSA (Multiple sequence alignment) を作成し、さらに自然言語モデルBERTにおけるMasked Language Modelのアイデアを転用してMSAへのマスク・変異導入を行います。
MSA (Multiple sequence alignment) の作成
ヒトもウマもカメも共通の祖先から分化して進化したために、ヒトが持っているタンパク質の多くはウマもカメも持っています。ヒトとウマとカメの外形的な見た目は全く異なる一方で、実はタンパク質レベルでの見た目であればそれほど変わりません。この傾向は下図に示すミオグロビン(酸素を運搬するヘモグロビンの仲間)のような生命維持に不可欠なタンパクであるほど顕著となります。
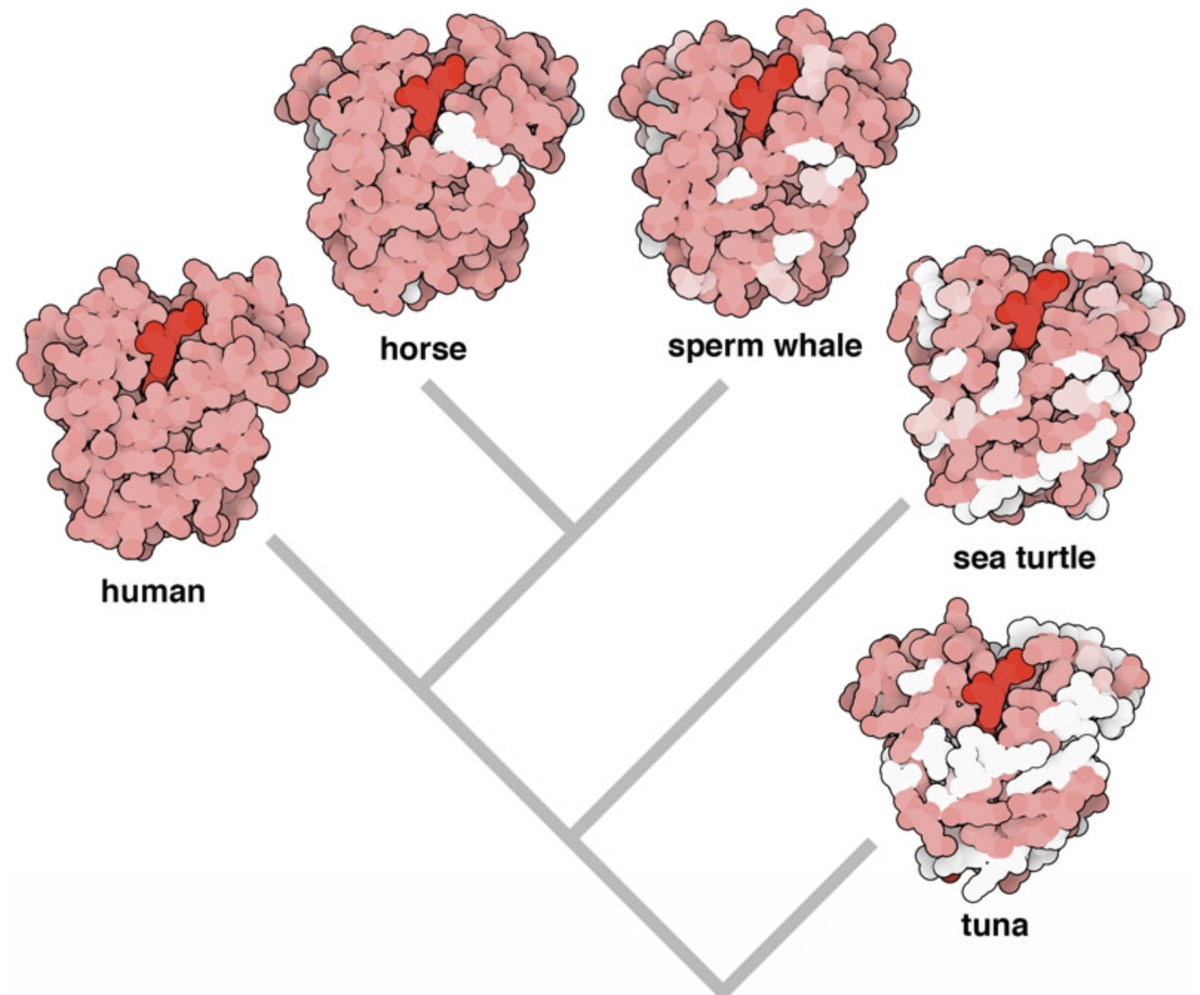
白色で表示されている部分はヒトのミオグロビンとは全く異なるアミノ酸が使われている
ここで重要なのは、同種のタンパク質であれば生物種が変わっても全体的な立体構造はあまり変わらないが、アミノ酸配列レベルではそれなりに差異が生じているということです。ゆえに、あるタンパク質についてさまざまな生物種のアミノ酸配列を並べたもの(=MSA)は、アミノ酸配列-タンパク質立体構造相関を考えるうえで重要な手掛かりとなります。
ただし、アミノ酸配列は分子進化の過程で変異するだけでなく長くなったり短くなったりするために妥当なアミノ酸配列の整列を得るのは容易なことではなく、バイオインフォマティクス分野ではより妥当なMSAを作成するための手法研究が古くから行われてきました。AlphaFold2が使用している HMMER は隠れマルコフモデルに基づくMSA作成手法の実装です。

なお、タンパク質立体構造解析に比べてタンパク質アミノ酸配列の解析は格段にコストが低いために、立体構造は不明だけどもアミノ酸配列ならわかっているというタンパク質が大量にあります。
MSA作成手順(概要):
- 入力配列をクエリとして配列の類似性スコアが一定値以上の最大5000配列でMSAを作成する
- 配列を雑に間引く(Supl. 1.2.6)
- 間引いてなお配列数が128以上の場合は、ランダムに128配列を選択する
配列数を128まで絞っているのは単純に計算コストの問題であり、深い意味はないことに留意ください。また、128配列に選ばれなかった配列についてそのまま捨ててしまうのはもったいないということで統計情報だけは利用するなどさまざまな工夫をしています(Supl. 1.2.7 MSA clustering)が、枝葉の処理なのでここでは詳細を割愛します。
MSAへのBERT風マスク導入
自然言語モデルBERTでは文章中の単語の15%程度をマスクトークンに置き換えて、この穴埋めクイズを事前学習(pre-training)として行うことにより言語への理解を獲得します。(Masked Language Model)
AlphaFold2でもMSAに対してBERTと同様なマスク置き換えを行い、MSA穴埋めクイズを通してAF2ネットワークにMSAの読み方を理解させることを目指します。

ただし、BERTとは異なりAF2では事前学習(MSA穴埋めクイズ)と目的タスク(立体構造予測)の訓練を分離せず、MSA穴埋め予測クイズのロス項と立体構造予測のロス項を足したものをトータルロスとしてまとめて学習します。(詳細は後述)。
テンプレート構造の検索(任意)
たとえばヒトのミオグロビンタンパクの立体構造を予測したいとします。ここで、もしヒトと十分に近縁であるサルのミオグロビンタンパクの立体構造が既知であるならば、サルのミオグロビンタンパクの立体構造をテンプレート(鋳型)としてヒトミオグロビンタンパクの立体構造を生成するだけで十分に品質の高い予測立体構造が得られます。
AlphaFold2においてもテンプレート構造情報を入力に含めることができますが、必須入力ではない上にablation study (論文 Fig. 4)より使わなくてもパフォーマンスがほぼ変わらないとされているので詳細説明は省略します。
1. Embeddingモジュール
このモジュールではスパースな入力値に対してEmbedding(活性化なし全結合層、つまり線形変換)を行うことでdenseなベクトルに変換します。さらに各入力データを統合し、MSA representation および Pair Representation を出力します。初見では複雑な処理に見えますが、必須のパス(任意パスは灰囲み)だけを見ればMSAに目的配列情報を付加する程度のごくシンプルな処理であることに気づきます。

入力データのOne-hot化
入力データはonehot化されています。自然言語処理に慣れた人にはonehot化からのembeddingはお馴染みの定型処理ですが、そうでない人のために何をやってるかの図を置いときます。ここでアミノ酸タイプとは、天然アミノ酸20種+残基不明+欠損+マスクトークンの23タイプです。

MSA Representation
MSA representaion は生のMSAに立体構造予測を行いたいアミノ酸配列(ターゲット配列)情報を紐づけたものと理解できます。また、直後にテンプレート立体構造のtorsion angleがconcatされることから側鎖レベルの詳細な立体構造情報を保持する役割も担っていると解釈できます。

Pair Representation(残基ペア表現)
Pair Representation(残基ペア表現)とは残基間の関係性(たとえば残基-残基タイプや残基間の空間的距離など)を表現することにより、主鎖レベルの大雑把な立体構造情報を保持する役割を担っていると解釈できます。初期状態では残基-残基タイプ程度の情報しか持ちませんが、AF2ネットワークを進むことにより残基間関係の情報が書き加えられていきます。たとえば、もし立体構造テンプレートを使用している場合は直後にテンプレート立体構造における残基間距離情報が追記(加算)されます。
2. Evoformerモジュール
Evoformerモジュールではself-attention機構によってMSA表現および残基ペア表現からの特徴量抽出を行います。やりたいことはTransformerのEncoder部とほぼ同じですが、MSAおよび空間グラフとしてのタンパク質の物理的制約を意識してやや変わったattention設計を行っています。また、MSA表現と残基ペア表現で情報交換が行われていることも特徴的なアーキテクチャです。

① axial-attentionによるMSA表現の特徴抽出
上図よりMSA表現からの特徴抽出パスには
- MSA row-wise gated self-attention with pair bias
- MSA column-wise gated self-attentio
- Transition (ただの全結合層)
の3つのブロックが存在することがわかります。
A. MSA row-wise gated self-attention with pair bias:

このブロックでは配列方向(row-wise)にaxial self-attention(軸方向注意)を適用することにより、配列内での情報交換を促進します。この(配列, row-wise)軸方向注意の仕組みは、ちょうど人間がMSAを配列方向に眺めて「このあたりはヘリックス*7構造っぽい」とか「システインが複数あるから分子内でスルフィド結合(S-S結合)を形成するかも」と考えつつMSAに注釈をつけていくようなプロセスを再現しています。
なお、このaxial attention(軸方向注意)はAF2論文での新規提案手法ではなく画像認識分野で提案された手法の転用となっています。

さてこのMSAについてのself-attentionですが、attention機構を見慣れた人であれば残基ペア表現(Pair representaion)をdot-product affinitiesにバイアスとして加算していることに強烈な違和感を覚えるのではないでしょうか。 安直には残基ペア表現をQuery, MSA表現をKey, Valueとしてattentionをしたくなります。しかし、残基ペア表現(Pair representaion)が保持している情報が残基-残基ペアの空間的関係性であることを思い起こせばバイアスとして加算することはごく自然な発想であることに気づきます。すなわち空間的に近い残基ペアについて大きなattention-weightsが割り当てられるようになるのです。
最後に目につくのはgatingですが、これはLSTMなどにおけるゲート機構と全く同じ役割を担っていると考えられます。すなわち不要な情報を削る”ゲート”の役割です。シグモイド関数で活性化されており素早く0→1を切り替えることができるために、効果的に情報の取捨選択を行えます。
B. MSA column-wise gated self-attention:
MSA row-wise gated self-attentionの軸を残基位置方向に変えただけでやってることはほぼ同じなので技術説明は割愛します。人間がMSAを残基位置方向に眺めて「この残基位置は生物種にわたって変異がほぼがないからきっと重要な残基なのだろう」とか「この残基位置はそれなりに変異あるけど疎水性アミノ酸ばかりだなあ」とか考えつつMSAに注釈をつけていくようなプロセスを再現しています。
② 残基ペア表現(Pair representation) の特徴抽出
残基ペア表現(Pair representation)は残基間の空間的な位置関係情報を保持するように設計されているために、 残基ペア表現の特徴抽出ブロックもまた残基間の空間的な位置関係に着目した設計となっています。

たとえば上図において残基i-k間の空間的距離と残基j-k間の空間的距離が決まったとすると、三角不等式より "残基i-j間の空間的距離 <= 残基i-k間の空間的距離 + 残基j-k間の空間的距離" でなければならないという制約が生じます。 言い換えると、残基i-j間のペア表現を更新するときには残基i-k間のペア表現および残基j-k間のペア表現と事前に情報交換を行う必要があるということです。関係者への根回しは大事。
そこでEvoformerでは関係の深い残基ペア表現間での情報交換の促進を
- Triangular multiplicative update
- Triangular self-attention
の2つのブロックで実現しています。ただし、あくまで残基間の物理的な制約を意識して設計されたブロックというだけであり、実際に物理的制約を満たすことは何ら保証されていないということには注意してください。
A. Triangular multiplicative update:
このブロックでは残基iとすべての残基間のペア表現(i行目)および残基jとすべての残基のペア表現(j行目)を使用して残基i-j間のペア表現を更新します。論文Fig3では3残基の組み合わせごとにfor文で処理するような印象を受けますが、Supl. Fig6では行ごとにまとめた計算効率の良い等価処理を説明しています。また、更新前の残基i-j間のペア表現がgating機構を通して情報の取捨選択をコントロールしていることがわかります。

ちなみにこのブロックはTriangular self-attentionの軽量versionとして設計されたものの、Triangular self-attentionと併用することによって精度向上することがわかったという開発秘話が論文Supl.に記述されています。
B. Triangular self-attention:
設計意図はTriangular multiplicative updateと全く同じですが、こちらのブロックではattentionが使用されています。論文Fig.3では残基iとすべての残基間のペア表現(i行)を使用して残基i-j間のペア表現を更新することをfor文で繰り返すような印象を受けますが、論文Supl.では残基iとすべての残基間のペア表現(i行)を使用して残基iとすべての残基間のペア表現(i行)を更新する、という計算効率のよい等価処理で説明されています。

3. 構造モジュール (Structure module)
構造モジュール (Structure module)では、Evoformerによって特徴抽出されたMSA表現と残基ペア表現と現在の主鎖立体構造
を入力として、各残基への「追加」の回転・並進指示
および 各残基のねじれ角
を出力します。※ i: iteration, r: 残基番号

ここで、"現在の主鎖立体構造"とは、(x, y, z)で表現されるようなリアルな座標ではなく、各残基についての原点からの回転・並進操作 T=(R, t)で表現されることに注意してください。回転・並進操作Tは各残基のCα(主鎖の中心炭素)に対して適用されます。
初めて構造モジュールに到達した場合は、"現在の立体構造"としてすべての残基が原点に集合する並進・回転操作 が初期立体構造として与えられます。なお、この構造初期化スキームは論文中でブラックホール初期化*8と呼称されています。
IPA(Invariant Point Attention)モジュール
構造モジュールではまずIPAモジュールによって、MSA表現と残基ペア表現、そして現在の主鎖立体構造 が統合されます。

IPAモジュールの上半分についてはEvoformerのMSA row-wise gated self-attention with pair biasとほぼ同じことをやってるだけなので説明を省省略します。
IPAモジュールの下半分では現在の残基間距離情報を取得する処理を行っています。このために残基数×p点の座標セットを生成し、現在の主鎖構造を表現している回転・並進操作Tを適用したうえで座標セット間の距離を算出しています。

Tを適用する前の座標セットに固定点を使うのではなくネットワークに動的生成させている意義はよくわかりません。残基タイプを考慮してネットワークがいい感じに座標セットを生成してくれる、たとえばアスパラギン酸のような細長い残基であれば細長い座標セットになるような効果があるのかもしれません。
座標点数pについて、query, keyについてはp=4座標点を生成しています。これは3点以下の座標点では回転操作の効果が薄れるためでしょう。valueについてはp=8座標点と多めに生成しており、より詳細な側鎖構造を意識している気がします。
・ Invariant(不変性)とは?
Invariant Point Attentionの「Invariant」とは、IPAモジュールの出力が主鎖構造のグローバルな回転・並進に依存しないよということを示しています。残基間の相対距離情報のみを利用しているのでグローバルな回転・並進操作に出力が依存しないのは直感的にも明らかです。※証明はSupl.1.8.2
(主鎖のグローバルな回転・並進操作: Pymolなどの3D分子Viewerで対象分子をグルグル回す操作と同様)
Predict relative rotations and translations(主鎖構造の更新)
IPAモジュールの出力するMSA表現は現在の立体構造情報を含むすべての情報を統合した最終MSA表現です。このブロックでは最終MSA表現から相対的な回転・並進指示を予測します。
とはいえやってることは全結合層で残基数分の回転行列と並進ベクトルを出力するだけです。ただし、回転行列(3×3)は直接予測するのではなくquaternion*9の予測を回転行列に変換します。quaternion知らなくともこれを使うと予測すべきパラメータが減って嬉しいくらいの理解でOK。

出力されるのは「相対的」な回転・並進操作Tなので、現在のTに出力されたTを適用することで現在のTを更新します。

各残基のねじれ角予測
Structureモジュールに突入する直前のMSA表現とIPAの出力した最終MSA表現から各残基の側鎖レベルの構造=ねじれ角 を予測します。
ω, Φ, Ψは主鎖のねじれ角(二面角)であり、χ1-4は側鎖のねじれ角です。たとえばグリシンなどは側鎖をもたないアミノ酸なのでχ1-4を利用しませんが深層学習の都合上で予測だけは行います。


最終MSA表現だけでなく、s_init = Structureモジュールに突入する直前のMSA表現 が併用されているのは、側鎖構造は主鎖構造に依存するので構造修正の手戻りが多いためでしょう。側鎖構造のバックアップを保持しているとも解釈できます。
なお、角度予測なので安直には[0, 2π]の範囲のスカラ値を予測したくなりますが、そうではなく2次元ベクトルを予測して回転行列に変換するほうが良いとのことです*10。
立体構造の出力
ここまでで主鎖構造(各残基の位置)と詳細構造(各残基のねじれ角)が決まったので一意的に立体構造を出力できます。しかしこれは暫定的な中間出力構造です。Structure moduleは8回繰り返すことで1サイクルとなっていますので、改善された主鎖立体構造と最終MSA出力を次ブロックの入力として同じ作業を繰り返しましょう。
最終的に出力された立体構造については軽量な分子シミュレーション(AMBER分子力場を使用した構造緩和)を行い、物理化学的に無理な構造を解消します。どの程度の無理があったかはfine-tuning時のみロス関数に使用します(後述)。
ロス関数
AF2のロス関数は複数種類のロス関数の重みづけ和をとったものとなっています。また、初期トレーニング時と自己蒸留データセットを用いたfine-tuning時(後述)ではロスの項が異なる(増える)ことに注意してください。これは物理化学的な無理(Lviol)のような細かい構造変化にセンシティブに反応する項を初期から考慮していると学習が不安定化するためであると考えられます。

Lfape:
最終出力された予測立体構造と正解立体構造とのズレの指標であるFAPEスコアに基づいたロス項。FAPEはRMSD(RMSD - Protein Data Bank Japan)スコアと似たような意味合いだが回転・並進ベクトルに基づくためキラリティを考慮しやすい。また、構造ズレが大きすぎる場合はClippingされる*11。
Laux:
8ブロックの繰り返しで構成される構造モジュールの、ブロックごとの中間予測構造についての平均簡易FAPEロス+ねじれ角予測ロス。簡易FAPEなのでCαについてのみ算出する。
Ldist:
残基間距離(distogram)の予測ロス。Lfapeと相関するはずだがこちらはより大雑把な構造ズレに特化しているので学習を安定させるのではないかと思われる。
Lmsa:
BERT風のマスクが適用されたMSAの穴埋めクイズを通じてAF2ネットワークにMSAの読み方を理解してもらうためのロス項。BERTのpre-trainingを同じ役割が期待されているはず。係数が大きいので実質的に事前学習(pre-training)っぽくなっているのでは。
Lconf:
モデルの信頼性予測スコアであるpLDDTの予測ロス。※後述
Lexp_resolved:
それが実験的に解かれた構造であるかの予測ロス?よく意味がわからない。
Lviol:
AMBER分子力場による構造緩和に基づく、立体衝突などの分子力学的な無理の大きさ。
「予測の信頼性」の予測:pLDDTスコア
AlphaFold2では「予測の信頼性(正確さ)」を予測することを目指します。
より具体的には最終予測立体構造と教師立体構造間の残基ごとに算出されるIDDT-Cαスコアを予測し、その予測誤差をネットワークのロス項に含めます(Lconf)。このIDDT-Cαスコアの予測値についてpLDDTと呼称します。
なおIDDT-Cαスコアとは2つのタンパク質立体構造のズレを妥当に算出するために設計されたスコアのようです。私は改造されたRMSD(RMSD - Protein Data Bank Japan)スコア程度の認識しかできていません。
pLDDTの定義から明らかなようにこのスコアに物理的な意味はありません。が、pLDDTは立体構造が既知の類似配列が乏しいような配列を予測する場合にはスコアが下がると思われますので予測信頼性の予測という表記通りの視点では一定の意味がありそうです。ただし、立体構造既知の類似配列に乏しい ∝ 定まった構造を取りにくいので立体構造が解かれてない ∝ 物理化学的に不安定、 のような疑似相関が見出される可能性は十分にあるかと思います。
pLDDTは後述する自己蒸留における、立体構造が既知の類似配列が乏しい配列についての予測立体構造を除外するような用途であれば存分にworkすることが期待できます。
更なる精度向上のためのトリック
Recycling
48のEvoformerブロックと8のStructureブロックを超えたあなたの手元には、十分に特徴抽出されたMSA表現とPair representaion、そしてすべての残基が原点に集合した初期構造と比較すればはるかに改善された主鎖立体構造を持っています。
ではこの3つを新たな入力として強くてニューゲームしましょう。再開始はEmbeddingモジュールの"R"と書いてある位置です。
論文Fig.4ではこのRecyclingによって構造精度が徐々に改善するタンパク(緑 T1064*12)があることが示されています。※48ブロックで1cycle

自己蒸留データセットによるFine-tuning
実験的に解かれたタンパク質立体構造データセット(PDBデータセット)よるトレーニングをある程度終えた後は、データセットを水増ししてfine-tuningを行います。
すなわち配列だけはわかっているタンパク質の立体構造を予測し、予測信頼性スコアが高い立体構造をデータセットに合流させることで教師立体構造データセットの大幅な増強(水増し)を実現します。
このようなラベルなしデータに予測ラベリング(疑似ラベルづけ)を行い、データセットを増強することで精度向上させるという手法は、画像分類タスクで提案された Noisy-studentの手法を踏襲したものとなっています。Noisy-studentは有名かつ汎用性が高い手法であるのでweb上に解説記事が多く存在するため、詳細説明は割愛します。
なお、予測構造のフィルターとなっている信頼性スコアには上述のpLDDTではなく残基間距離予測に基づいた信頼性スコアを使用しているのですが、これは「その時点でpLDDTが開発されてなかっただけであり、もしplDDT使ってたとしても同じような結果になると思うよ」、という記述があります。
まとめ
AlphaFold2は構造生物学のドメイン知識に基づいたコンセプトをディープラーニングで超人化することに成功した手法と言えます。ドメインの伝統的な発想を高度な深層学習エンジニアリング力で超人化するというDeepMindのアプローチは機械式時計のような精密工芸品を眺めている気持ちになります。こういうドメイン知識を活用するアプローチはAlphaZeroでも成功していますしDeepMindの勝ちパターンになってますね。
本記事の記述に間違いを見つけたらコメントにて指摘をお願い致します。
補足情報
数値のカテゴリ表現
AF2では残基間距離のように数値表現が必要な値は基本的にカテゴリ表現にエンコードします。
本ブログ別記事の転用ですが、下図を見れば数値をカテゴリ表現するという処理の意味がつかめるかと思います。
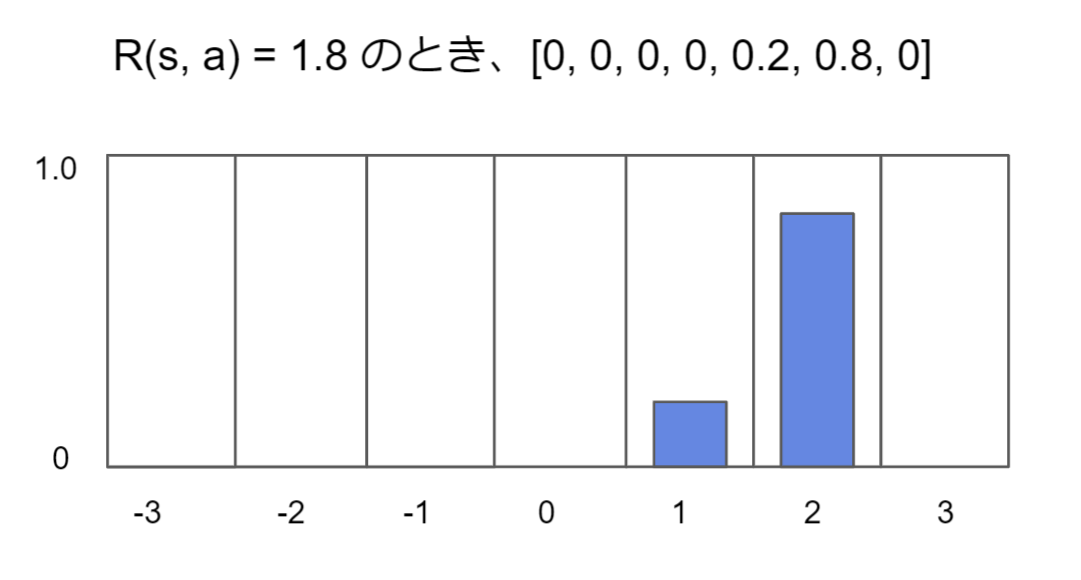
数値をカテゴリ表現にすることにより
・ スケール感が揃うのでネットワークにやさしい
・ 数値予測時にはロス関数にクロスエントロピーが使えて学習が安定する
・ 複数のロス項間のスケール感の差を気にしなくて良いので学習が安定する
というようなメリットがあります。
このトリックはMuZero(AlphaZeroの後継手法)でも多用されています。
MuZeroの実装解説(for Breaktout) - どこから見てもメンダコ
*1:新たに立体構造が解明された未発表タンパク質が問題として出題される
*3:膜タンパクの話はややこしくなるのでNG
*4:分子シャペロンの話はややこしくなるのでNG
*5:ただし論文内でモジュールと呼称されているのはEvoformerとStructure moduleのみ
*6:論文ではdata pipelineの呼称
*7:らせん型の部分立体構造
*8:中心にブラックホールでも存在しないと原子が激しく反発してはじけ飛ぶため
*9:3D-CGを扱うときは必須の概念
*10:Supl. Table 2の直後あたりとAlgorithm 25
*11:極端なHuber lossみたいなものと理解している